Working with ipyparallel
In this example we want to use basico from ipython, in a ipyparallel, as the normal approach with using multiprocessing does not seem to work across operating systems within the jupyter notebooks (read: i could not make it work on Windows).
This means, that to run this example, you will need the ipyparallel package installed, which can be achieved by uncommenting the folling line:
[1]:
#!pip install -q ipyparallel
once that is installed, you will to switch to the home screen of the jupyter notebook, and start the cluster manually:

otherwise this notebook will fail with an error like:
---------------------------------------------------------------------------
FileNotFoundError Traceback (most recent call last)
Cell In[3], line 1
----> 1 cluster = ipp.Cluster.from_file()
2 rc = cluster.connect_client_sync()
3 # wait to get the engines and print their id
File ~/env/lib/python3.11/site-packages/ipyparallel/cluster/cluster.py:556, in Cluster.from_file(cls, cluster_file, profile, profile_dir, cluster_id, **kwargs)
554 # ensure from_file preserves cluster_file, even if it moved
555 kwargs.setdefault("cluster_file", cluster_file)
--> 556 with open(cluster_file) as f:
557 return cls.from_dict(json.load(f), **kwargs)
FileNotFoundError: [Errno 2] No such file or directory:
'~/.ipython/profile_default/security/cluster-.json'
Once the ipyparallel is installed and the cluster is running continue as follows:
[2]:
from basico import *
import ipyparallel as ipp
we could create a cluster manually, in my case i just created one in the cluster menu in the jupyter setup, so to access it we just need to run:
[3]:
cluster = ipp.Cluster.from_file()
rc = cluster.connect_client_sync()
# wait to get the engines and print their id
rc.wait_for_engines(6); rc.ids
[3]:
[0, 1, 2, 3, 4, 5, 6, 7]
Now we create a direct view with all of the workers:
[4]:
dview = rc[:]
The model we use is the BioModel 68, since we will need the model many times, i download it once and save it to a local file:
[5]:
m = load_biomodel(68)
save_model('bm68.cps', model=m)
remove_datamodel(m)
And define the worker method, the worker just loads a biomodel (in case it was not loaded into the worker before, otherwise it accesses the current model). It then chooses a random initial concentration for 2 species, and computes the steady state of the model. Finally the initial concentrations used and the fluxes computed are return. In case a seed is specified, it will be used to initialize the rng:
[6]:
def worker_method(seed=None):
import basico
import random
if seed is not None:
random.seed(seed)
if basico.get_num_loaded_models() == 0:
m = basico.load_model('bm68.cps')
else:
m = basico.get_current_model()
# we sample the model as described in Mendes (2009)
cysteine = 0.3 * 10 ** random.uniform(0, 3)
adomed = random.uniform(0, 100)
# set the sampled initial concentration.
basico.set_species('Cysteine', initial_concentration=cysteine, model=m)
basico.set_species('S-adenosylmethionine', initial_concentration=adomed, model=m)
# compute the steady state
_ = basico.run_steadystate(model=m)
# retrieve the current flux values
fluxes = basico.get_reactions(model=m).flux
# and return as tuple
return (cysteine, adomed, fluxes[1], fluxes[2])
We can invoke this method synchronusly in the notebook to obtain the result:
[7]:
worker_method()
[7]:
(1.5236813977490542,
29.805870872058716,
0.04158430196391732,
0.9584156980360827)
now we want to do that many times over (passing along the index to set the seed every time to get about the same result on my machine):
[8]:
%%time
sync_results = []
for i in range(2000):
sync_results.append(worker_method(i))
CPU times: user 2.06 s, sys: 55 ms, total: 2.12 s
Wall time: 2.08 s
here a utility method plotting the result:
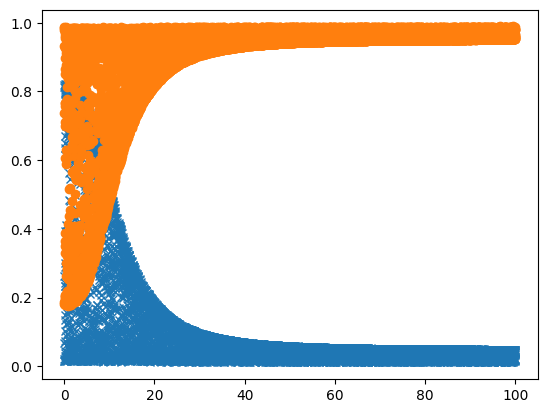
[9]:
def plot_result(results):
cys = [x[0] for x in results]
ado = [x[1] for x in results]
y1 = [x[2] for x in results]
y2 = [x[3] for x in results]
plt.plot(ado, y1, 'x')
plt.plot(ado, y2, 'o')
plt.show()
[10]:
plot_result(sync_results)
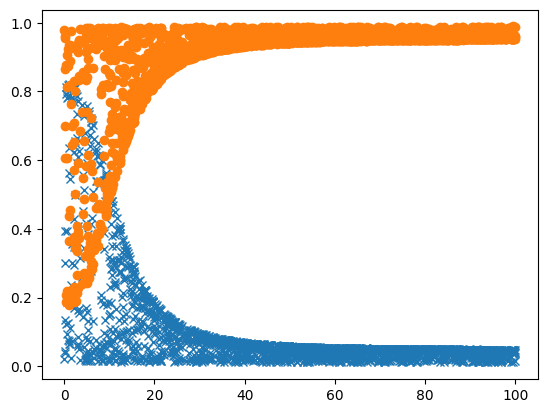
So that for me took some 10 seconds (even though the model was loaded already), so the next thing to do is to run it in parallel. Here i start 10000 runs, passing along the seed for each one, to make it reproducible:
[11]:
%%time
ar_map = dview.map_async(worker_method, [i for i in range(10000)])
ar_map.wait_for_output()
CPU times: user 412 ms, sys: 259 ms, total: 671 ms
Wall time: 3.72 s
[11]:
True
And now we can plot that as well:
[12]:
plot_result(ar_map.get())